

Fibrose cística: o que é, causas, sintomas e tratamentos

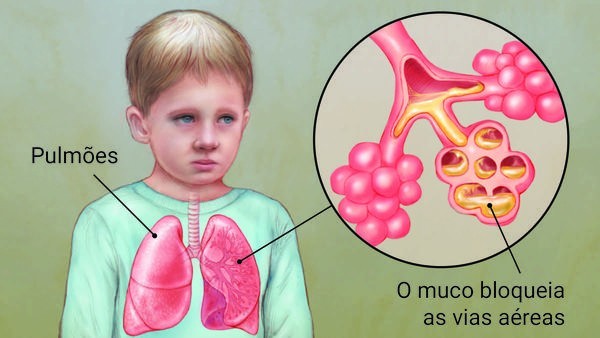
A fibrose cística é uma doença genética, crônica, que afeta principalmente os pulmões, pâncreas e o sistema digestivo. É uma doença pouco conhecida por grande parte da população, entretanto, a Fibrose Cística é muito perigosa e pode provocar danos sérios a saúde.


Atinge cerca de 70 mil pessoas em todo mundo, e é a doença genética grave mais comum da infância. Um gene defeituoso e a proteína produzida por ele fazem com que o corpo produza muco de 30 a 60 vezes mais espesso que o usual.
O muco espesso leva ao acúmulo de bactéria e germes nas vias respiratórias, podendo causar inchaço, inflamações e infecções como pneumonia e bronquite, trazendo danos aos pulmões.
Esse muco também pode bloquear o trato digestório e o pâncreas, o que impede que enzimas digestivas cheguem ao intestino. O corpo precisa dessas enzimas para digerir e aproveitar os nutrientes dos alimentos, essencial para o desenvolvimento e saúde do ser humano.
Pessoas com fibrose cística frequentemente precisam repor essas enzimas através de medicamentos tomados junto às refeições, como forma de auxílio na digestão e nutrição apropriadas.
O que é a fibrose cística?
A fibrose cística, também conhecida como Mucoviscidose, é uma doença genética autossômica (não ligada ao cromossoma x) recessiva (que são necessários para se manifestar mutações nos 2 cromossomas do par afetado) causada por um distúrbio nas secreções de algumas glândulas, nomeadamente as glândulas exócrinas (glândulas produtoras de muco).
O cromossoma afetado é o cromossoma 7, sendo este responsável pela produção de uma proteína que vai regular a passagem de cloro e de sódio pelas membranas celulares.
A proteína afetada vais ser a CFTR (regulador de condutância transmembranar). E tal como a proteína, o próprio canal de cloro vai sofrer uma mutação do qual vai resultar um transporte anormal de iões de cloro através dos ductos das células sudoríparas e da superfície epitelial das células da mucosa.
Vai ocorrer então uma alteração no transporte dos iões de cloro através das glândulas exócrinas apicais, resultando dessa anormalidade, uma permeabilidade diminuída ao cloro, fazendo com que o muco da fibrose cística fique cerca de 30 a 60 vezes mais viscoso. A água por sua vez, como vai seguir o movimento do sódio de volta ao interior da célula, vai provocar um ressecamento do fluído extracelular que se encontra no interior do ducto da glândula exócrina.
Como detectar a fibrose cística:
Para diagnosticar a FC, atualmente são empregados os seguintes exames:
- Tripsina Imunorreativa (IRT): Para diagnóstico pré-natal, com teste de “Screenning”, realizado junto com o teste do pezinho.
- Teste do Suor: Medição dos níveis de íons de sódio (Na+) e cloro (Cl-). É rápido e indolor.
- Coprológico Funcional: Análise das fezes. Exame complementar.
- Pesquisa Genética: Análise do DNA, detectando as mutações para a FC.
Nunca será demais bater na tecla diagnóstico precoce. Uma vez observada a doença logo nos primeiros dias de vida da criança, as chances de aumentar a sua sobrevida serão muito maiores, assim como a fase de adaptação a doença transcorrerá de forma menos traumática para a criança e também para seus familiares.
Sintomas da fibrose cística:
Os sintomas da doença costumam variar de acordo com a idade do paciente. Os recém-nascidos e os mais novos costumam ter sintomas diferentes dos outros pacientes. Os recém-nascidos tem como principais sintomas:
- Dificuldade para ganhar peso;
- Desidratação sem motivo aparente;
- Secreções que atrapalham o funcionamento do intestino;
- tosse com secreção.
Pacientes mais velhos:
- Diabetes;
- Infecções respiratórias;
- Atraso na puberdade;
- Infertilidade (acredita-se que apenas 30 ou 40% das mulheres não conseguem engravidar e só
- 2% dos homens conseguem se tornar pais biológicos);
- Sinusite crônica;
- Perda de peso;
- Diarreia crônica;
- Deformidade nos dedos e unhas;
- Desnutrição;
- Formação de pólipos nasais;
- Cirrose biliar.


Diagnóstico da fibrose cística:
Exames de triagem para a fibrose cística são realizados em todos os recém-nascidos nos Estados Unidos. Além disso, uma pequena gota de sangue é coletada em um pedaço de papel filtro e o nível de tripsina (uma enzima digestiva) é medido.
Se o nível de tripsina no sangue estiver elevado, os recém-nascidos passam por exames confirmatórios. Além disso, os exames confirmatórios incluem exames de suor e/ou genéticos. Mais de 90% dos novos casos de fibrose cística agora são identificados por exames de triagem em recém-nascidos.
Exames de portador podem ser realizados nas pessoas que querem ter filhos no futuro ou estão buscando receber cuidados pré-natais. Além disso, particularmente, os familiares de uma pessoa com fibrose cística podem desejar saber se têm maior risco de ter filhos com a doença.
Visto que a fibrose cística pode afetar vários órgãos, outros exames podem ser úteis. Uma vez que as concentrações de enzimas pancreáticas estão reduzidas, uma análise das fezes da pessoa pode revelar concentrações baixas ou não detectáveis das enzimas digestivas elastase, tripsina e quimotripsina (secretadas pelo pâncreas) ou concentrações altas de gordura. Além disso, exames de sangue são realizados para determinar se a secreção de insulina está baixa e se os níveis de açúcar no sangue estão elevados.
Testes de função pulmonar ( Testes de função pulmonar (TFP)) podem revelar um comprometimento da respiração e são bons indicadores de como os pulmões estão funcionando. Além disso, radiografias e tomografia computadorizada (TC) do tórax podem ajudar a documentar infecções pulmonares e a abrangência dos danos no pulmão. Além disso, a TC dos seios nasais é realizada para pessoas que apresentam sintomas sinusais sérios, principalmente se elas têm pólipos nasais.
Causas da fibrose cística:
A fibrose cística é causada por um gene defeituoso que faz com que o corpo produza um líquido anormalmente denso e pegajoso, conhecido popularmente como muco, que se acumula nas passagens respiratórias dos pulmões e também no pâncreas.
Esse amontoado de muco resulta em infecções pulmonares que podem colocar a vida do paciente em risco, e podem levar a problemas digestivos graves também. A doença ainda pode afetar as glândulas sudoríparas e o sistema reprodutivo masculino.
A maioria das crianças com fibrose cística é diagnosticada até os dois anos de idade. Além disso, um número menor, no entanto, só é diagnosticado com 18 anos ou mais. Esses pacientes geralmente têm uma forma mais branda da doença.
Tratamentos:
O tratamento é multidisciplinar, pois envolve médicos, enfermeiros, fisioterapeutas e assistentes sociais e tem o objetivo de melhorar a sobrevivência e a qualidade de vida. “O principal foco na criança pequena é a nutrição adequada.
Seguir dietas mais calóricas e fazer reposição das enzimas do pâncreas pode ajudar o paciente a não sofrer de desnutrição, que está associada à piora da doença pulmonar. Quanto mais desnutrido estiver o paciente, pior para o pulmão”, diz o especialista.
Para melhorar o trato respiratório são realizados tratamentos para fluidificar as secreções, com uso de nebulizações, fisioterapia respiratória, que ajuda a expelir a secreção, e o uso de antibióticos inalatórios.
De acordo com o pneumologista, a fibrose cística não tem cura e o tratamento ajuda a retardar a progressão da doença. “Conforme a doença pulmonar evolui e o paciente apresenta dilatação dos brônquios e acúmulo de secreção, pode ocorrer insuficiência respiratória crônica e limitação das atividades diárias em casos muito avançados, o transplante de pulmão é a melhor opção e possui resultados satisfatórios”, conta Silva Filho.
Com os tratamentos atuais, os pacientes possuem expectativa de vida mais longa. Além disso, eles podem ter uma vida normal desde que realizem o tratamento adequado. Porém com o passar dos anos, a doença pulmonar pode piorar e a pessoa ficar incapacitada.
Em países de primeiro mundo, a sobrevida média é superior a 40 anos, daí a importância do diagnóstico precoce. Além disso, quanto mais cedo descobrir a fibrose cística, melhor a qualidade de vida e maior o tempo de sobrevivência do paciente.

Recomendações:
Procure afastar qualquer indício de sentimento de culpa se seu filho for portador. Além disso, você é tão culpado por ter-lhe transmitido esse gene quanto por ter transmitido o gene que lhe deu a cor dos olhos ou o tom da pele;
Verifique se o teste do pezinho inclui a triagem para a fibrose cística, quando nascer uma criança em sua família. Além disso, esteja atento à boa nutrição do portador de fibrose cística. Paciente bem nutrido pode atingir as curvas normais de peso e altura;
Convença o paciente a praticar atividade física. Natação, por exemplo, ajuda a trabalhar a musculatura da caixa torácica. Não se esqueça de que os portadores da doença, desde que adequadamente tratados, conseguem levar vida normal e de boa qualidade.